Alpers-Huttenlocher Syndrome
- aka Alper's Disease
- Considered a Neurological disorder
- Identified as a Progressive and severe encephalopathy with epilepsy
- Potential liver failure
- Autosomal Recessive Disorder
- Usually identified when individuals are between 2-4 years old however some individuals do not present symptoms until they reach the ages from 17-24
- Result of disease is often fatal
The disease occurs in about 1/100,000 to 1/250,000 individuals. Approximately 80% present symptoms within the first 2 years, and 20% present between 2 and 25 years of age. Individuals with this disease often show symptoms of motor dysfunction, seizures, cortical blindness, liver dysfunction, deafness, uncontrollable jerky movements and a often a degeneration of knowledge and higher thinking. There are many variation of the symptoms of Alpers-Huttenocher syndrome, some symptoms can be more severe or be more progressive while others do not. It's important to note that currently the cause of the large variation is unknown but is currently a point of research. The diseases symptoms are a consequence of dysfunction in the mitochondria, the energy center of the cell. The gene that causes this mitochondrial dysfunction and Alpers-Huttenlocher syndrome is called POLG. The gene provides instructions to make the alpha subunit, that is a part of polymerase gamma protein. Polymerase gamma functions in mitochondria, mitochondria are structures found in cells that use oxygen to convert the energy from food into food that the cell can use. Polymerase gamma is used to "read" sequences of mitochondrial DNA (mtDNA) and uses them as templates to produce new copies of mtDNA through mtDNA replication.
Alpers-Huttenlocher syndrome is caused by mutations found in the POLG gene. The most common mutation found in this syndrome is an amino acid change from an alanine(A) to a threonine(T), that makes up the the alpha subunit of polymerase gamma. These changes can result in a mutated polymerase gamma that becomes inefficient in its ability to replicate mtDNA. The A467T (467 is the position of the change) mutation is also common in other POLG-related disorders. The different types of disorders could be due to the other POLG gene found on the other chromosome. The majority of the POLG mutations involve the amino acid parts that make up the the alpha subunit of polymerase gamma. Mutations in this gene have been shown to produce a reduced number of copies of mtDNA in brain, muscle and liver cells. The depletion of mtDNA leads to a decrease in cellular energy, which is a possible contributor to the symptoms of Alpers-Huttenlocher syndrome., but there are still mutation combinations that can cause some of the other disorders, however not all of these combinations are known and still many are unaccounted for.
The majority of the POLG mutations involve the amino acid parts that make up the the alpha subunit of polymerase gamma. Mutations in this gene have been shown to produce a reduced number of copies of mtDNA in brain, muscle and liver cells. The depletion of mtDNA leads to a decrease in cellular energy, which is a possible contributor to the symptoms of Alpers-Huttenlocher syndrome.
Alpers-Huttenlocher syndrome is caused by mutations found in the POLG gene. The most common mutation found in this syndrome is an amino acid change from an alanine(A) to a threonine(T), that makes up the the alpha subunit of polymerase gamma. These changes can result in a mutated polymerase gamma that becomes inefficient in its ability to replicate mtDNA. The A467T (467 is the position of the change) mutation is also common in other POLG-related disorders. The different types of disorders could be due to the other POLG gene found on the other chromosome. The majority of the POLG mutations involve the amino acid parts that make up the the alpha subunit of polymerase gamma. Mutations in this gene have been shown to produce a reduced number of copies of mtDNA in brain, muscle and liver cells. The depletion of mtDNA leads to a decrease in cellular energy, which is a possible contributor to the symptoms of Alpers-Huttenlocher syndrome., but there are still mutation combinations that can cause some of the other disorders, however not all of these combinations are known and still many are unaccounted for.
The majority of the POLG mutations involve the amino acid parts that make up the the alpha subunit of polymerase gamma. Mutations in this gene have been shown to produce a reduced number of copies of mtDNA in brain, muscle and liver cells. The depletion of mtDNA leads to a decrease in cellular energy, which is a possible contributor to the symptoms of Alpers-Huttenlocher syndrome.
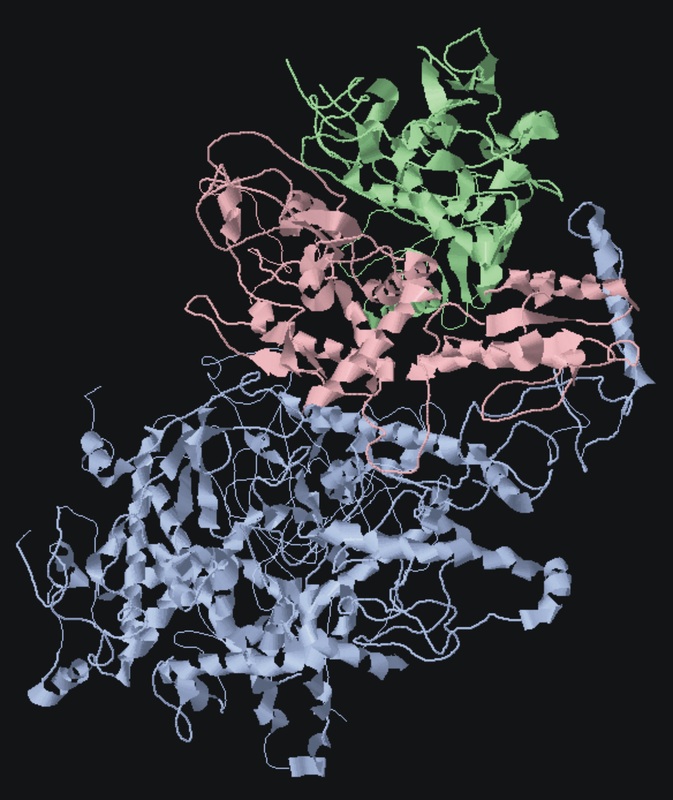
Polymerase gamma pictured above, the alpha subunit shown in blue which is affected by POLG mutations, the red and green are the other accessory subunits
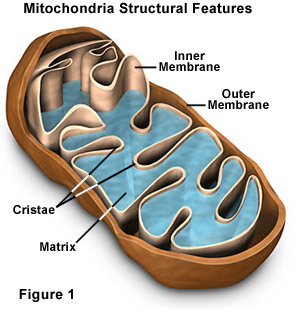
Alpers-Huttenlocher Syndrome has been identified as causing problems in the Mitochondria. The mitochondria helps produce energy for the cell and the disease directly affects the mitochondria's ability to efficiently produce energy for the cell.
Many symptoms are rarely seen at first in individuals early on, and the symptoms don't occur in a particular order in individuals and this makes diagnosis difficult until the disease has progressed. Brain MRI is initially normal but it can eventually show cerebral volume loss.
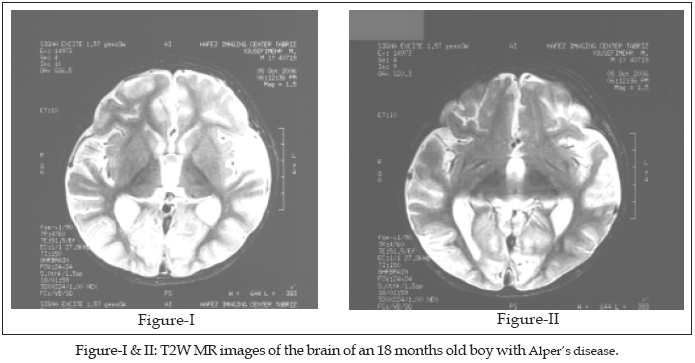
The brain imaging pictured above shows widespread cerebral atrophy in grey matter, as well as a decrease in white matter in the brain. There is also cortical thinning in the frontal, posterior temporal and occipital lobes. Grey matter is generally used to process information in the brain as well as cognition. White matter actively affects how the brain learns and functions. Above you can also see the occipital lobe atrophy is also visible in these scans.
Autosomal Dominant Inheritance Pattern
|
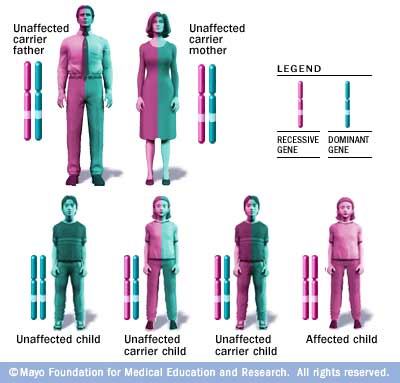
Alpers-Huttenlocher syndrome is inherited via a recessive inheritance pattern. This disorder is not sex-linked (disease not carried on X or Y chromosome) and males and females alike can develop this disease.This means that in order for an individual to be affected by the disease they need two copies of the diseased allele. If a child carries only one allele that carries the disease along with a normal allele, then the child will not be affected by the disorder. The picture below shows how this type of inheritance works.
|
Treatment
Currently there is no cure for Alpers-Huttenlocher disease and no way to slow its progression. Treatment revolves around treating the symptoms and general support of the victim. Anticonvulsants can be prescribed for the seizures however often the anticonvulsants don't work to treat the episodes. The pros/cons of using pills to sedate patients in order to treat the seizures have to be weighed against the actual effectiveness of the treatments due to the high ineffectiveness of the anticonvulsants. Physical therapy is also used to help relieve spasticity and maintain or increase muscle tone for patients.
Prognosis
Currently the prognosis for the disease is very poor most individuals die within the first decade of life. The seizures can become continuous with one leading to another which can often lead to death. Liver failure and cardiorespiratory failure due to nerve, brain and spinal cord interaction may also occur in individuals. Some however do live to there mid 30s but they often experience severe neurological deficiencies.
References
- "POLG Gene." Genetics Home Reference. US National Library of Medicine, June 2011. Web. 25 Mar. 2015.
- DiMauro, Salvatore. Mitochondrial DNA Deletion Syndromes. U.S. National Library of Medicine, n.d. Web. 19 Mar. 2015.
- Naiviaux, Robert, Pr. "The Portal for Rare Diseases and Orphan Drugs." Orphanet: Alpers Syndrome. N.p., July 2006. Web. 26 Mar. 2015.
- Tzoulis, C., Tran, G. T., Coxhead, J., Bertelsen, B., Lilleng, P. K., Balafkan, N., Payne, B., Miletic, H., Chinnery, P. F. and Bindoff, L. A. (2014), Molecular pathogenesis of polymerase gamma–related neurodegeneration. Ann Neurol., 76: 66–81. doi: 10.1002/ana.24185
Aaron Burd, [email protected], Last updated: 3/25/15 http://genetics564.weebly.com/
